
計算機シミュレーションやバイオインフォマティクス的手法を用いて、生体高分子の機能発現機構を研究しています。特に、蛋白質と核酸の複合体に注目し、転写因子やヌクレオソーム、複製・修復関連因子などがどのようにして分子を認識しているか、分子レベルで明らかにする研究を行っています。また、量子ビーム(X線、中性子線、電子線)を利用して得られた生体高分子の立体構造情報をもとにして、生体超分子の全原子座標構造モデルの構築や反応遷移状態の原子構造モデルを構築する方法論の開発を行っています。その他、蛋白質と核酸の分子認識における溶媒効果の研究、コヒーレントX線による単粒子の立体構造決定アルゴリズムの開発を行っています。
Latest news
第56回日本生物物理学会年会(岡山大学)2日目(9月16日)に、ワークショップ「マルチスケール・フィジクスで見えてくる生体高分子のダイナミクスと機能機序」を開催します。ここを参照(18ページ目)[PDFファイル/1.21MB]
新学術領域 クロマチン潜在能(平成30-34年度)が新たに発足しました。領域研究に参画しています。
ここを参照
Recent Publications
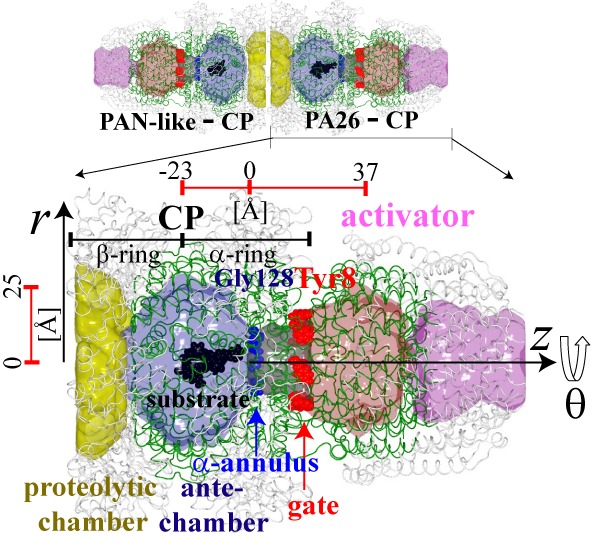
Ishida, H. (2014) Essential function of the N-termini tails of the proteasome for the gating mechanism revealed by molecular dynamics simulations , Proteins, 82, 1985-1999.
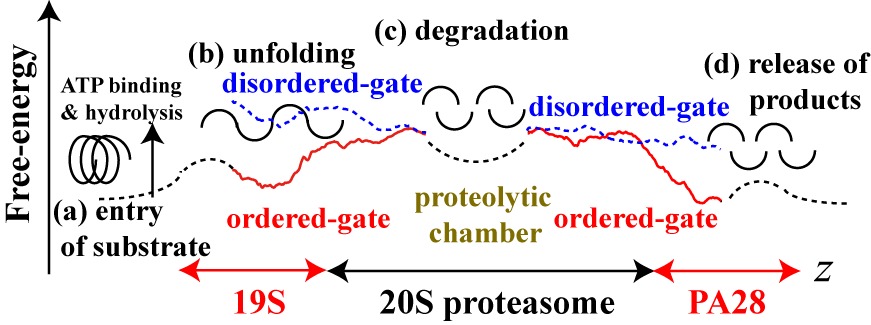
巨大な酵素複合体であるプロテアソーム(左上図)は、生体内でできた異常タンパク質や役目を終えて不要になったタンパク質を分解・排除します。更には、プロテアソームは活性調節因子と結合して様々な生命現象(癌、免疫、DNA修復、神経、老化)において中心的な役割を果たしています。Adaptively Biased Molecular Dynamics (ABMD)自由エネルギー計算を用いることにより、タンパク質分解酵素プロテアソーム内部を通過する基質移動の反応遷移状態を観測し、自由エネルギー地形を計算しました。本解析により、免疫反応における抗原ペプチド生成・排出に関与するハイブリッドプロテアソームの機能発現モデルを提唱しました(左下図)。
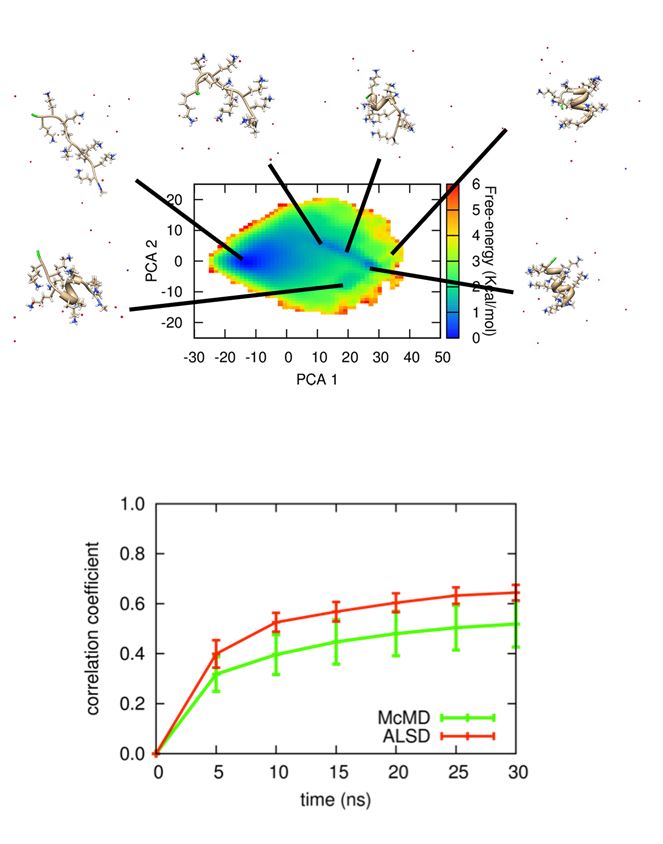
Ikebe, J., Sakuraba, S. and Kono, H. (2014) Adaptive lambda square dynamics simulation: An efficient conformational sampling method for biomolecules J Comput. Chem., 35, 39-50.
蛋白質やDNAは、それぞれアミノ酸やヌクレオチドが鎖状に連なった生体分子です。この鎖状分子は、低い自由エネルギーを持つ特定の立体構造に折り畳まれることによってその機能を発揮します。分子動力学シミュレーションで生体分子の自由エネルギー地形を再現することは、生物学の大きな目標のひとつです。しかし、従来のシミュレーション手法でこれを実現するためには莫大な時間を要します。そこで私たちは従来の手法よりも高速に生体高分子の自由エネルギー地形を再現することのできる手法、Adaptive Lambda Square Dynamics (ALSD)法を開発しました。
ALSDは、生体分子のポテンシャルエネルギー変化を促進させることによって、様々な立体構造を高速に探索することができます。私たちは、ポリリジン9残基ペプチドのテスト計算を行うことによって、ALSDは従来手法(McMD)よりも高速に、このペプチドの自由エネルギー地形を再現できることを確認しました。
(左上図)主成分解析(PCA)法によって描かれたポリリジン9残基ペプチドの自由エネルギー地形と代表構造。
(左下図)十分に長時間のシミュレーションから得られた上図の自由エネルギー地形と、短時間シミュレーションから得られた自由エネルギー地形の間の相関係数。
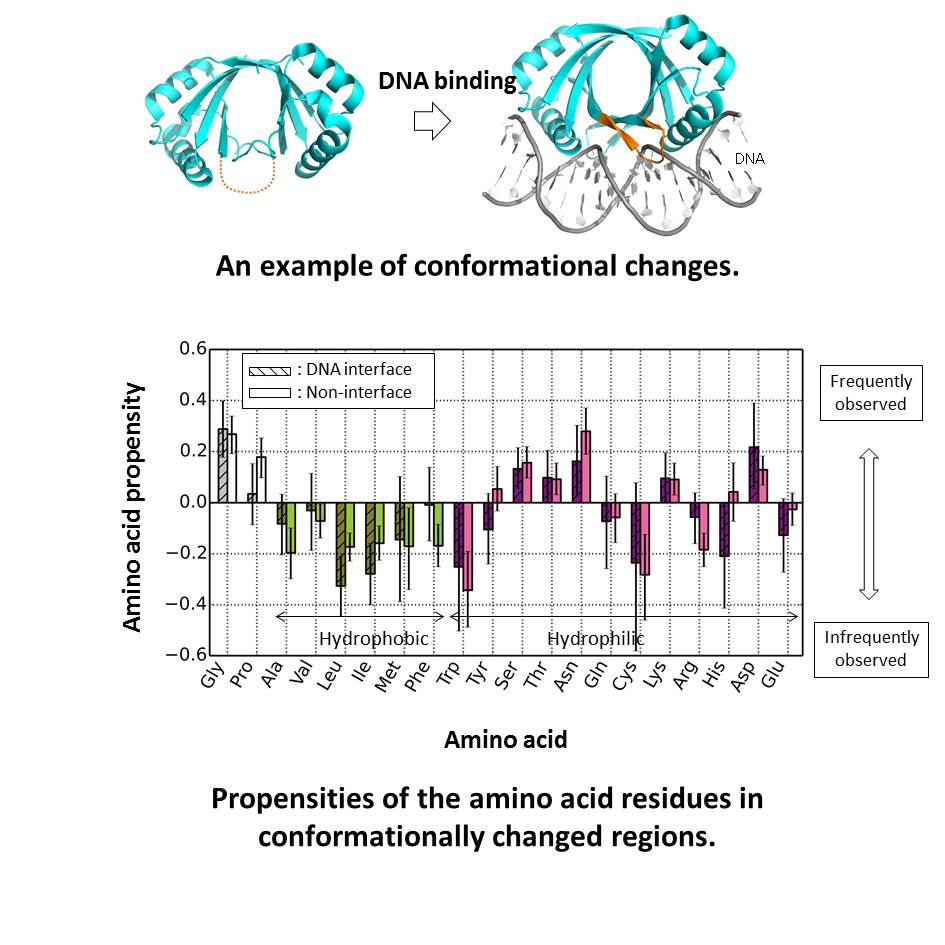
Sunami, T. and Kono, H. (2013) Local conformational changes in the DNA interfaces of proteins. PLoS One. 2013;8(2):e56080.
私たちの体内では、外界の状況に応じてタンパク質の合成が常に行われています。これは、細胞核内のDNAの特定の領域に、転写因子と呼ばれるDNA結合タンパク質が結合することによって始まります。このようなDNA結合タンパク質の中には、ステロイド受容体など多くの核内受容体が含まれています。これら受容体とDNAとの相互作用を薬剤によって促進・抑制することができれば、病気の治療薬として役に立つと考えられています。そのため、DNA結合タンパク質がどのように特定のDNAを認識するかその分子機構を明らかにすることが非常に大切です。
これまでに、個別のDNA結合タンパク質に関しては、X線結晶構造解析によって、DNAの認識機構が原子レベルで明らかにされてきました。しかしながら、その一般的な法則は、まだ十分に明らかになっているとは言えません。
私たちは、多くのX線解析データを注意深く観察し、DNA結合タンパク質がDNAと結合する際には、タンパク質に構造変化が生じていることを観察しました。そこで、私たちはタンパク質がDNA結合によってどのような構造変化を生じるのかを調べるために、構造アルファベットという方法でタンパク質構造を記述し、タンパク質の三次元構造の変化を統計的に解析しました。
その結果、まず、DNAが結合するタンパク質領域にはもともと構造変化しやすい領域が多く存在しており、DNAとの相互作用によって、様々な構造を生じていることを明らかにしました。次に、このような領域は、疎水的なアミノ酸が少なく、親水的なアミノ酸の一部や、グリシンやプロリンが多いなど、アミノ酸組成が特徴的であることを明らかにしました。DNAと接している領域が構造変化に適したアミノ酸組成を持っていることは、DNA結合タンパク質は、DNAの構造に合わせるために、構造変化しやすい性質を進化の過程で獲得してきたことを示しています。
タンパク質とDNAの分子認識は、生物の基本的な仕組みの一つです。私たちは、構造変化に注目することによって、その仕組みの一部を明らかにすることに成功しました。
![]()
PDF形式のファイルをご覧いただく場合には、Adobe社が提供するAdobe Readerが必要です。
Adobe Readerをお持ちでない方は、バナーのリンク先からダウンロードしてください。(無料)
Adobe Reader provided by Adobe is required to view PDF format files.