
学会GMP基準についての解説
1.定義
学会製造基準で使用される用語の定義が記載されています。
- 1.1「PET薬剤」とは
- 1.2「バッチ」とは
- 1.3「サブバッチ」とは
- 1.4「ロット」とは
- 1.5「バリデーション」とは
- 1.6「ベリフィケーション」とは
- 1.7「クオリフィケーション」とは
- 1.8「出荷」とは
2.PET薬剤製造部門及びPET薬剤品質部門
PET薬剤製造施設には、PET薬剤の製造管理に係る部門(「製造部門」)と品質管理に係る部門(「品質部門」)を設置し、それぞれ独立していなければならない、と規定されています。
<PET薬剤製造施設の体制(組織図例)>
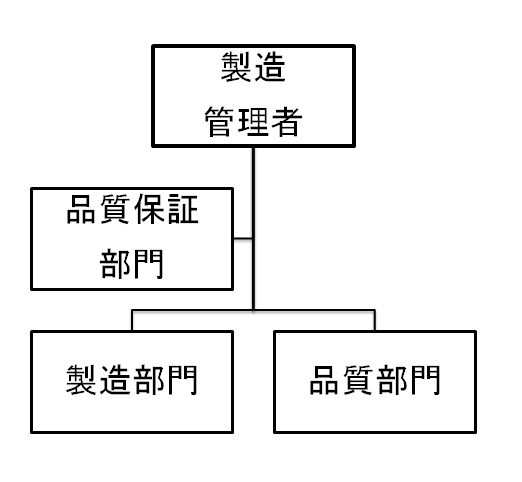
(注)品質保証部門を体制上設置することが難しい場合は、品質部門が品質保証機能を担っても良いとされています。
3.PET薬剤の出荷の管理
「出荷」とは、PET薬剤製造エリアから払いだすことを指しています。本基準は、原則として院内使用を対象として規定されています。
出荷されたPET薬剤はヒトに投与されますので、非常に重要なプロセスです。出荷可否決定や出荷の手順、出荷可否決定者の要件等を規定しています。
4.PET薬剤に関する文書
いわゆる「製品標準書」について記載されています。PET薬剤の製造に必要な情報として、リファレンスとして必要な項目を規定しています。
5.手順書等
PET薬剤製造施設の管理に必要となる、基準書・手順書について、製造施設ごとに設置するよう記載されています。文例集では、これに従い「一般管理基準書」「衛生管理基準書」「製造管理基準書」「品質管理基準書」を作成しています。
6.PET薬剤の製造管理
製造管理に関する事項(主に製造管理基準書に記載)が記載されています。
6.1.1「製造指図書」
PET薬剤の製造指図及び製造手順を具体的に記述した文書です。
6.1.3「製造記録書」
製造指図書に基づき正しく製造・工程管理が実行されたことを証明するための記録書ですので、紙、もしくは電子署名等を使用した改ざん不可能な形態の電子ファイルで記録する必要があります。
6.1.4「表示及び包装の管理」
手順どおりにPET薬剤が製造されたとしても、包装表示(ラベル)が間違っていては、目的のPET薬剤であることは全く保証されません。ラベルの作製、容器へのラベルの貼付及び包装作業等についても、手順及び記録を作成することが必要です。
6.1.5「原材料、PET薬剤及び資材(PET薬剤の容器)の管理」
PET薬剤及び原材料、資材については、試験検査を実施後、きちんと出納管理や保管管理を行う必要があります。試験検査後に貼付する承認ラベル(適合/不適合ラベル)は、製造施設認証監査でも必ずチェックするポイントとなります。
6.1.6「構造設備の清浄」
6.1.7「職員の衛生管理」
文例集では、いずれも衛生管理基準書に記載されています。
6.1.8「構造設備のバリデーション及びクオリフィケーション」
製造管理に使用する構造設備、機器等の設置や一定使用期間後、必ず適格性を確認する必要があります。
6.1.9「構造設備の管理」
製造管理に使用する構造設備、機器等は、使用時点検や校正及び保守・点検を記録し、保管する必要があります。
6.1.10「清浄管理区域と無菌操作区域の管理」
環境モニタリングについて規定する項目です。文例集では、衛生管理基準書に記載されています。
6.1.11「PET薬剤の製造管理の品質部門への報告」
製造記録や受入試験記録、衛生管理記録等は、適切に製造管理が行われているか、品質部門(品質保証機能)による確認が必要とされます。各記録書には品質部門の確認印欄(サイン欄)を設けましょう。
6.1.12「その他必要な業務」
職員以外の外部者の立入に関する制限について記載されています。外部者の立入に際しても、入退室記録や教育訓練の記録が必要になります。
6.2「交差汚染」
交差汚染の危険があるPET薬剤製造施設では、原料やPET薬剤の交差汚染が発生しないよう、手順書を作成したり、洗浄バリデーション等で予め清浄であることを担保しておく必要があります。
7.PET薬剤の品質管理
品質管理に関する事項(主に品質管理基準書に記載)が記載されています。
7.1.1、7.1.2「検体採取と試験検査」
検体採取に関しても、手順書を作成し、保管管理の記録を残す必要があります。採取した検体の試験検査結果については、生データも含めて記録・保管しますが、感熱記録紙の場合は印字が消えてしまう可能性があるため、コピーしたものを保管することも可能です。
7.1.5「委託製造」
PET薬剤の製造工程(全部または一部)を外部に委託することを指します。外部業者からオペレーターが派遣され、自施設内で製造工程作業にあたる場合は該当しません。この場合には、派遣オペレーターは自施設内で職員登録を行ってください。
7.1.6「出荷判定基準の適合と出荷」
出荷可否決定に際しては、品質試験結果のみならず製造管理記録(製造記録や施設・職員の衛生管理記録や環境モニタリング記録等)の確認も必要です。不適合の場合、再試験の手順に従って1回のみ再試験の実施が可能です。
7.1.7「安定性試験」
新規PET薬剤で学会製造施設認証監査を行う場合には、安定性試験の手順作成や実施を求められることがあります。PET薬剤合成装置メーカーで安定性試験が実施され公開されている場合には、各PET薬剤製造施設で安定性試験を実施する必要はありません。
7.1.8「参考品の保存」
PET薬剤に関しては、参考品として保存可能な量が少なく、原則として必要とされる2回分の試験に必要な量を確保することは困難なため、投与と試験検査に供した残りを保存する形で構いません。参考品の出納管理も忘れず実施しましょう。
7.1.9「設備及び機器のバリデーション及びクオリフィケーション」
品質管理に使用する構造設備、機器等を設置する際は、必ず適格性を確認する必要があります。
7.1.10「試験検査設備及び機器の管理」
品質管理に使用する構造設備、機器等は、使用時点検や校正及び保守・点検を記録し、保管する必要があります。
7.1.11「外部試験検査機関等での試験検査の実施」
残留溶媒測定等、品質試験の全部もしくは一部を外部へ委託する場合の、試験検査記録書が必要です。外部業者からオペレーターが派遣され、自施設内で品質試験検査にあたる場合は該当しません。 この場合には、派遣オペレーターは自施設内で職員登録を行ってください。
8.外部試験検査機関等の利用
試験検査等を外部へ委託する場合、試験検査依頼項目や検体の輸送方法等、あらかじめ確認及び取り決めておく必要があります。
9.バリデーション及びベリフィケーション
- バリデーションとは、製造工程や試験検査法等の規定されたプロセスが予定通りの結果をもたらすことを、その変動要因を考慮した上で検証する事です。例えば、製造工程であれば、その工程により基準通りの生成物が製造できる事を予め繰り返し確認しておくことになりますし、試験法の場合では、測定対象物の標準品を使用し、その試験法の精度や再現性が基準通りであることを確認します。
- ベリフィケーションは、新しいプロセスを用いて得られた結果が適合しているか、品質部門の品質保証機能が承認することで信頼性を担保します。
10.変更の管理
製造管理及び品質管理に関する変更は、全て手順書に従って管理しなければなりません。特にPET薬剤の品質に影響を与える可能性がある変更に関しては、必要に応じて事前評価を行います。
11.逸脱の管理
逸脱事例が生じた場合には、ただちに軽重を判断する必要があります。普段より発生しうる逸脱事例を分類分けしておくと良いでしょう。
12.品質等に関する情報及び品質不良等の処理
いわゆるクレーム処理に該当します。PET薬剤の出荷後に品質等に関する情報が入った場合には、PET薬剤との因果関係の有無に関わらず、全て記録します。情報がきちんと入手できるよう、日頃からPET薬剤使用部門との連携体制を構築しておきましょう。
13.回収処理
PET薬剤の品質等の問題で回収が必要となった場合、手順に従い回収処理を行います。回収したPET薬剤は他のものと区別して保管し、回収に至った原因究明や改善措置が採られた後、適切に処理しましょう。
14.自己点検
PET薬剤製造施設の自己点検作業は、年に1回以上の頻度で実施します。学会が作成している監査チェックシートを利用すると便利です。
15.教育訓練
PET薬剤製造施設で作業を行う職員(派遣を含む)や外部立入者全てに対し、教育訓練を実施しなければなりません。それぞれの技量に応じた教育訓練を実施し、所定の訓練を修了した者だけが作業を行う事ができます。
必要な資格認定として、学会が指定しているのは無菌操作認定のみですが、各施設で必要に応じ、その他の資格認定を設ける事も可能です。
16.文書及び記録の管理
基準書や手順書等の作成の仕方から記録の保管管理まで、文書管理の手順書に記載しておきます。廃止文書は、電子媒体も含めて廃止後5年間は保管が必要です。
17.PET薬剤の製造施設の構造設備
無菌操作区域でグレードA管理の装置を使用するため、適切なグレード管理が可能な構造設備を構築する必要があります。